

Translate this page into:
Validation – In pharmaceutical industry: Equipment validation: A brief review

-
Received: ,
Accepted: ,
How to cite this article: Jindal D, Kaur H, Patil RK, Patil HC. Validation – In pharmaceutical industry: Equipment validation: A brief review. Adesh Univ J Med Sci Res 2020;2(2):94-8.
Abstract
Validation is the procedure which authorizing documentary evidences that prove, the following process/ method or activity will consistently produce the product which leads to the expected result (predetermined requirements). The validation program in pharmaceutical industries involves various components which are related to processing, cleaning, facilities, equipment, or instrumentation. In this review article, we will go through a brief discussion about one of the most preferred method of validation which is equipment validation. In equipment validation, we will discuss about its types in detail, what kind of documentation is required and applications/importance of validation in pharmaceutical industry. Nowadays, equipment validation becomes the regulatory requirement for pharmaceutical companies to precede the validation of new equipment’s/instruments. Meanwhile, the process of validation requires detailed knowledge of that instrument which is going to validated; therefore, the validation is usually performed by the company which supply that equipment.
Keywords
Validation
Documentary
Evidences
Equipment
Pharmaceutical Industries

INTRODUCTION
The validation process is the documented evidence which provides a high degree of assurance to a desired result with predermined compliance. The term validation is widely used in pharmaceutical industries. This term comes from the word “valid or validity” which means “legally defined”. The validation concept was first proposed by the Food and Drug Administration (FAD) in the mid-1970s to improve the quality of pharmaceutical products. Since a wide variety of procedures, methods or activates are validated to check and improve their quality.[1-3]
Types of validation [Figure 1]
Validation is divided into following subsections which include:[3]
Analytical method validation
Process validation
Cleaning validation
Equipment validation
Let’s take an overview of different types of the validation process and discuss in detail about equipment validation and its phase with their importance in pharmaceutical industries.
-
Analytical method validation: The purpose of analytical validation is to verify that the selected analytical procedure will give reliable results that are adequate for the intended purpose. There are different parameters which come under analytical method validation. These are as follows:[2,4]
Accuracy
Precision
Repeatability
Reproducibility
Specification
Linearity
Range
Detection limit
Quantitation limit
-
Process validation: This type of validation demonstrates documented proves, which carries a higher degree of surety that the process will consistently produce a product which meets all the predetermined quality characteristics and specifications. The process validation also assures the repeatability of the process and decreases the risk of manufacturing problems which lead to an increase in output of predetermined quality.
On the bases of the stage of production under process validation, it can be of four types which are as follow:
Prospective validation
Concurrent validation
Retro specific validation
Revalidation.
Cleaning validation: Cleaning validation provides documented set up with a high degree of surety that particular system/equipment or part of equipment is consistently clean-up to predetermined quality and acceptable limits. Pharmaceutical products are contaminated by variety of substances such as lubricants, airborne materials, prepared product residues, and microbes. Hence, an adequate cleaning procedure plays an important role to prevent contamination and cross contamination.[1,5]
Equipment validation: Equipment validation is established documented set up that proves any equipment works correctly and leads to accepted and accurate results (predetermined result). The process of equipment validation is based on the principle that equipment must be designed, constructed, maintained, and adapted to perform the operations which are to be carried out. Equipment’s are the basic component of pharma industries; therefore, before performing a process in pharma industries, it becomes primary important to issue equipment validation (documented evidences of equipment).[5,6]
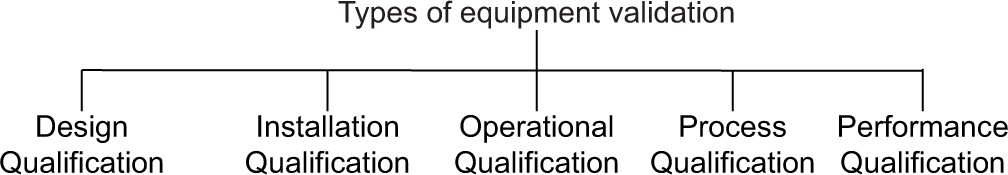
Types of equipment validation [Figure 2]: The process of equipment validation is not a single step activity that it has different phases which have further subsections or steps, these are as follow:[6]
Design qualification
Installation qualification
Operational qualification
Performance qualification
Process qualification
Types of equipment validation
The process of pharmaceutical equipment validation in pharma industries is quite simple to proceed. The various stages of the process are thoroughly investigated and documented in accordance with approval from pharmaindustry/company. The process of procurement normally starts by the production of required documentation and user requirement specification (URS). To perform validation project/plan (VP), a form of change request (CR) should be taken from the existing facilities. As earlier the management agreed to proceed, the request is issued to perform validation project (VP). Then with approved VP, the validation protocol can be started that required to verify that all the requirements documented in the URS and all cGMP requirements are fulfilled.
Phases of equipment validation [Figure 3]: The process of equipment validation is mainly divided into three phases:[6,7]
Phase – 1: Pre-validation phase.
Phase – 2: Process validation phase.
Phase – 3: Validation maintenance phase.
Pre-validation phase
-
Design Qualification (DQ): It is a documented verification of design of the equipment and manufacturing facilities. The main purpose of Design qualification is to make sure that all the requirements for the systems should clearly defined at the start. Design qualification process will illustrate that all quality aspects are fully considered at the design stage. It defines the functional and operational specifications of the instrument with all requirements, as mentioned in the user requirement specification (URS) and the applicable cGMP rules and regulations. The accomplishment of documented qualification must verify that the given design will follow:[8]
User requirement specification ( URS)
Functional specification (FS)
Tender specification and drawing
Purchase specification
Vendor qualification
-
User requirement specification (URS): It includes the list of requirements/expectations of the customer in the equipment. The general customer requirements are as follows:
Size of equipment and space occupied by it.
Effectiveness and durability of the equipment.
Working speed of the equipment.
Equipment should be with low noise and air pollution.
Availability of the spare parts and also provide services at minimal cost.
Overall good construction.
Installation Qualifications (IQ): Installation qualification confirms that the précised equipment has been received and installed as per target and agreement in exact design or format in the undamaged form with parts, spares, services gauges, and other required compounds. It is documental verification of that the equipment has been installed and calibrated appropriately. The purpose of IQ is to ensure that all the aspects of the equipment are installed correctly match with the original (URS) design. As per the manufacture’s recommendations for installation, the working sites working environmental conditions are documented and confirmed that they are suitable for the operation of the instrument.[9]
The documentation of installation includes:
Details of supplier and manufacture.
Equipment name, color, model and serial number.
Date of installation and calibration.
Process validation phase
• Operational Qualifications: Operational qualification ensures that installed equipment/instrument will function perfectly according to its operation specification in the mention environmental conditions. It also checks that the equipment function perfectly to meet pre-assigned performance criteria and ensure how the testing results are recorded. The purpose of the operational qualification is to make sure that all the dynamic conditions well comply with original (URS) design. For verification, it includes traceable electric stimulators and standards which verify that equipment is processing correctly as required. Operational qualification gave high degree of assurance that the equipment functionally verifies compliance of manufactures specifications and user required specifications (URS). Operational qualification is also known as process validation that it ensures the processing of the equipment from the user and manufacturer point of view with proper documentation verification.[10]
Documentation for operational validation includes:
Finalized and approved operations (functions testing)
Certified calibrations • System stability test results
Applications of S.O.P.s
Performance Qualification: Performance qualification ensures that the equipment consistently performs functions according to the mentioned specification which appropriates to its daily/routine use. It is a documented verification process which verifies that all aspects of facility, utility, and performance of equipment meeting pre-assigned acceptance criteria from user requirement specification (URS) and manufactures specifications. Performance qualification is performed under controlled conditions that are similar to daily sample analysis and it is performed on daily basis (at least repeated after a week) when equipment is used or functioning performed. It is also known as system suitability testing, its testing frequency is quite higher than that of operational qualification. The test frequency depends not only on functioning of equipment but also on the stability of each unit of entire system which contributes to the analysis result.[11]
Documentation for performance validation includes:
Performance qualification report
Process stability testing reports (long-term productivity)
Acceptance of the product record (costumers reviews)
Actual product and process parameters documentations.
Routinely performed test results documentation.
Re-validation: The performance of re-validation is done when the operating equipment and system have been modified in some ways due to any reason. Revalidation of the equipment is very helpful in maintaining the validation status of the equipment and entire system which work as a unit. The process of revalidation is also used for the periodic checking of the validation as per the government guidelines.[4,5,12]
Re-validation is further divided as follows:
Periodic/scheduled re-validation
Re-validation after change/modifications
Periodic re-validation process refers to the re-validation process which carried out in pharmaceutical industry at periodic intervals and it is mandatory especially when the company made any change in the formulas, procedures, manufacturing systems, packaging, and support system such as electricity/ power supply, water supply, and steam. A separate and well qualified team will come for the process of re-validation in case of equipment re-validation that the analyst will come from the manufacturer side. Minor change in the product may affect the product’s quality up to a great extent hence to carry validation become necessary even after the minute change. Sometimes operational and performance tests were re-performed, which were done even during first time validation.
- Types of validation. Design Qualification
- Types of equipment validation.

- Phases of equipment validation.
The following is the changes for that re-validation is necessary these are as follow:
Change in raw material.
Change in manufacturing process.
Change in equipment/system.
Change in supporting systems.
Change in packaging materials.
Validation maintenance phase
Maintenance qualification (MQ): Maintenance qualification will review and verify the acceptability of the maintenance controls to confirm the equipment/ system integrity. Maintaining requires a documented periodic review of processes and system/equipment. It is a periodic process which ensures that the equipment should not affect the safety, quality, and strength, identity of the manufactured product either through its contamination or structure. The process of maintenance qualification includes routine servicing and necessary repairs.[13]
The documentation for maintenance qualification includes:
outine services records
Maintenance contracts details
List of authorized services engineers
Application of Equipment validation: The following is the importance of equipment validation in pharmaceutical industries.[14]
Validation of equipment reduces costs by reducing rejects, reworks, and downtime.
Decrease the risk of non-compliance regulatory.
High rate of customer satisfaction.
Analytical tests methods and calibrations are proceeded.
It also reduces testing in in-process and final product.
Also improve employee’s awareness.
Make maintenance of equipment easier.
Give more rapid and reliable start-up for new equipment’s.
Help in development of validation master plan for the facility.
The validation documentation can be used as a presentation in case of inspection. (As a legal proof).
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Validation in Pharmaceutical Industry (6th ed). United Kingdom: Informa Healthcare Publication; 2016. p. :51-70.
- [Google Scholar]
- Good Manufacturing Practices for Pharmaceuticals (5th ed). New York: Marcel Dekker Publication; 1997. p. :66-80.
- [Google Scholar]
- The Pharmaceutical Master Validation Plan: The Ultimate Guide to FDA, GMP, and GLP Compliance Hardcover-Illustrated. Cleveland: CRC Press;. ;2001:115-25.
- [CrossRef] [Google Scholar]
- An overview of pharmaceutical validation. Quality assurance view point. Int J Res Pharm Chem. 2011;1:1003-14.
- [Google Scholar]
- Guidance on validation and qualification of processes and operations involving radiopharmaceuticals. EJNMMI Radiopharm Chem. 2017;2:8.
- [CrossRef] [PubMed] [Google Scholar]
- Equipment and its Qualification, Rutendo Kuwana Technical Officer Geneva: World Health Organization; 1992. p. :14-96.
- [Google Scholar]
- Guidance for Industry, Process Validation: General Principles and Practices Washington, DC: U.S Food and Drug Administration; 2009.
- [Google Scholar]
- Validation of Compendia Methods Rockville, MD: United States Pharmacopeia and National Formulary. The United States Pharmacopoeia Convention Inc.; 1995. p. :1612-710.
- [Google Scholar]
- A history of validation in the United States, Part I. Pharm Technol. 1991;15:82-96.
- [Google Scholar]
- Validation Guidelines for Pharmaceutical Dosage Form Canada: Health Products and Food Branch Inspectorate; 2009. p. :8-13.
- [Google Scholar]
- An approach to the characterization and technology transfer of solid dosage form processes. Pharm Technol. 1982;6:139-56.
- [Google Scholar]
- An Overview: Pharmaceutical Validation. Int J Innovative Pharm Sci Res. 2014;2:2476-97.
- [Google Scholar]
- Pharmaceutical Process Validation: An Overview. Int J Appl Pharm Biol Res. 2014;3:243-62.
- [Google Scholar]
- Validation Theory and its Application Laguna: Telstar Manufacturing Corporation; 2009. p. :12-6.
- [Google Scholar]




