

Translate this page into:
Homology modeling of actin protein in Leishmania donovani and in silico prediction of active drugs

*Corresponding author: Dimpal Rani Bansal, Department of Pharmaceutical Sciences, Adesh Institute of Pharmacy and Biomedical Sciences, Adesh University, Bathinda, Punjab, India. dimpalbansal@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Bansal DR, Patil HC, Patil RK. Homology modeling of actin protein in Leishmania donovani and in silico prediction of active drugs. Adesh Univ J Med Sci Res 2020;2(1):52-7.
Abstract
Objectives:
Leishmaniasis is a disease caused by leishmania parasite which is genus of trypanosome protozoa. Leishmania donovani promastigote inhibits biogenesis of phagolysosome due to the accumulation of periphagosomal F-actin. This inhibition of phagosome maturation gives favorable environment for differentiation of promastigote-to-amastigote and causes disease progression. L. donovani actin (LdACT) has been found to have unconventional biochemical behavior due to the different amino acid region in its sequence suggesting that it must have a three-dimensional (3D) structure different from eukaryotic actins making it a more specific for predication of antileishmanial drugs which is main objective of this study.
Material and Methods:
For carrying out this study, protein sequence was retrieved from the database SWISSPROT, analyzed by BIOEDIT software followed by primary and secondary structure prediction by PROTPARAM and SOPMA. A 3D structure of same was constructed by homology modeling using the yeast actin-human gelsolin segment 1 complex (protein data bank [PDB] ID:1yag) as a template with the help of Swiss model. The final model obtained was further accessed by PROCHECK and VERIFY 3D software which ensured the reliability of the model. This model of actin protein was further used for screening different chemical compounds with high binding affinity by GOLD and DISCOVERY STUDIO.
Results:
The results give information about the some inhibitors having highest binding affinity to the actin protein.
Conclusion:
This study will be useful for the development of pharmacophore models for in silico predication of active drugs as a part of antileishmanial drug therapy.
Keywords
Homology modeling
Leishmania donovani actin
Protein-protein docking
Modeller 9v8
GOLD
Discovery studio

INTRODUCTION
Leishmania are the protozoan parasites and the members of the family Trypanosomatidae, which are unicellular organisms with the presence of a single flagellum and a DNA-rich, mitochondria- like organelle, the kinetoplast. The various species of Leishmania cause diseases termed leishmaniasis that affect the people worldwide.[1] These parasites circulate among vertebrates, and transmitted by dipterid phlebotomine flies of the genera Phlebotomus and Lutzomyia.[2]
For the development of new antiparasitic chemotherapy, which is effective, we use the application of in silico drug designing which is based on structure-based drug design and ligand-based drug design uses protein structure predication and screening of chemical databases by computational methods as an inexpensive tool to identify potential chemotherapeutic agents.
Actin has emerged as an attractive target for structure based on drug design against Leishmania donovani.[3] Actin protein, acts as cytoskeleton protein which is responsible for the cell morphology and acts as the building block required for cell motility, contracting ability and for intracellular transportation in eukaryotic cells.[4-6] Furthermore, actin is found in the nucleus where it is responsible for playing an important role in chromatin remodeling,[7,8] and other functions such as RNA processing, transport, and other similar processes.[9,10] Hence, the actin is a multifunctional which associates with microtubules formation during processes, such as cell migration, and vesicle transport,[11,12] and also with the plasma membrane regions that are involved during endocytosis.[13,14] Actin protein has different role in cell multiplication, growth, and development.[5] L. donovani actin shows unique biochemical properties that have not been found for any other actin till date so it is novel form of actin. The mammalian actin and Leishmania actin show differences in the amino acid sequences of which are, namely, aa 40–53, aa 194–200, and aa 266–281. Hence, we assume that we can use L. donovani actin as an good target for in silico designing novel antileishmanial drugs.[15]
In this study, we are targeting actin protein of L. donovani as a drug target. As the structure of the protein is not available so model protein is constructed by homology modeling and the protein-ligand interaction studies will be carried out to find out the potential inhibitors.
MATERIAL AND METHODS
The are different tools, which are used for searching journals, databases, for the analysis of protein, for retrieving sequence of protein and also comparison of the sequences, build protein model, validation of model, visualization of three-dimensional (3D) structure, active site finding, protein-ligand interaction, ligand design, and ADMET prediction are the following
Sequence retrieval, template selection, and alignment of sequences
National Centre for Biotechnology Information (www.ncbi.nlm.nih.gov/) was used for the retrieval of amino acid sequence of actin protein from protein database.[16] After that the search for comparative modeling starts. For this searching, the PDB of known protein structures and using the target sequence as the query. FASTA and BLAST are the popular program for comparative study.[17] After obtaining a list of potential templates using searching methods, then we select one or more templates that are good for the particular modeling of protein. As the overall sequence similarity to the target increases, the quality of template also increases. Moreover, this quality if template is decreases with the number and length of gaps in the alignment. The simplest rule for selection of template is to select the structure which has the highest sequence similarity to the modeled sequence. Clustal W used for alignment of the sequence of template and target proteins.[18,19]
Protein modeling and validation of models
SWISS-MODEL server was used for the comparative modeling of 3D protein structures.[20] Further discovery studio was used for refinement of the model. The web based program SOPMA was used for the estimation of secondary structure of the protein.[21] RasMol is the visualization tool for the protein structure. ProtParam is a tool from ExPASy which is used for the computation of various physical and chemical parameters for a given protein. The computed physical and chemical parameters include the molecular weight, theoretical PI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity. Ramachandran map is used for the validation of the model which is carried out after the refinement process and calculation is computed with the PROCHECK program. It is used to checks the stereochemical quality of a protein structure on the phenomena based on analyzes residual geometry and overall structure geometry.[22] Finally, the validated models were subjected to further protein ligand docking studies and other calculations.[23]
Docking studies
A docking studies assess the ability of the actin protein homology model to use in the in silico drug designing due to its ability to binding small molecule ligands to the appropriate target binding site. PubChem database was used to retrieve 3D structures of the compounds as a SD file format. A library of chemical compounds having proved actin inhibitor activity such as oxazole ring system, hydrazide, sulfonamide, macrolide, and some miscellaneous compounds were generated and all these compounds were then subjected to docking to find out the best scoring ligands. For designing of chemical library, ACDChemSkech11.0 is used. It is a drawing package that allows you to draw chemical structures and Docking of protein model and ligand was performed using AccelrysDS2.1 and Gold 4.0.[24]
RESULTS
Template selection and alignment of sequence
Template selection of actin protein from L. donovani (LdAct) was done by BLAST analysis against PDB proteins and this template selection is based on the highest sequence similarity as mentioned above. Moreover, highest results belonged to actin protein from structure of the yeast actin-human gelsolin segment 1 complex (PDB Id: 1YAG Identities (72%). ClustalW program was used for sequence alignment which is a program for multiple sequence alignment for DNA or proteins. Multiple sequence alignments of actin protein with template 1YAG by ClustalW are done in [Figure 1].

- Sequence alignment of actin protein with template 1YAG by Clustal W.
Protein modeling and validation of models
A 3D model of protein was constructed with the Swiss model on the basis of its template structure. Moreover, after construction of model protein, we done the refinement of model by discovery studio and validation by PROCHECK program was also done. Using this process, the best pharmacophore model of L. donovani actin is obtained in [Figure 2].
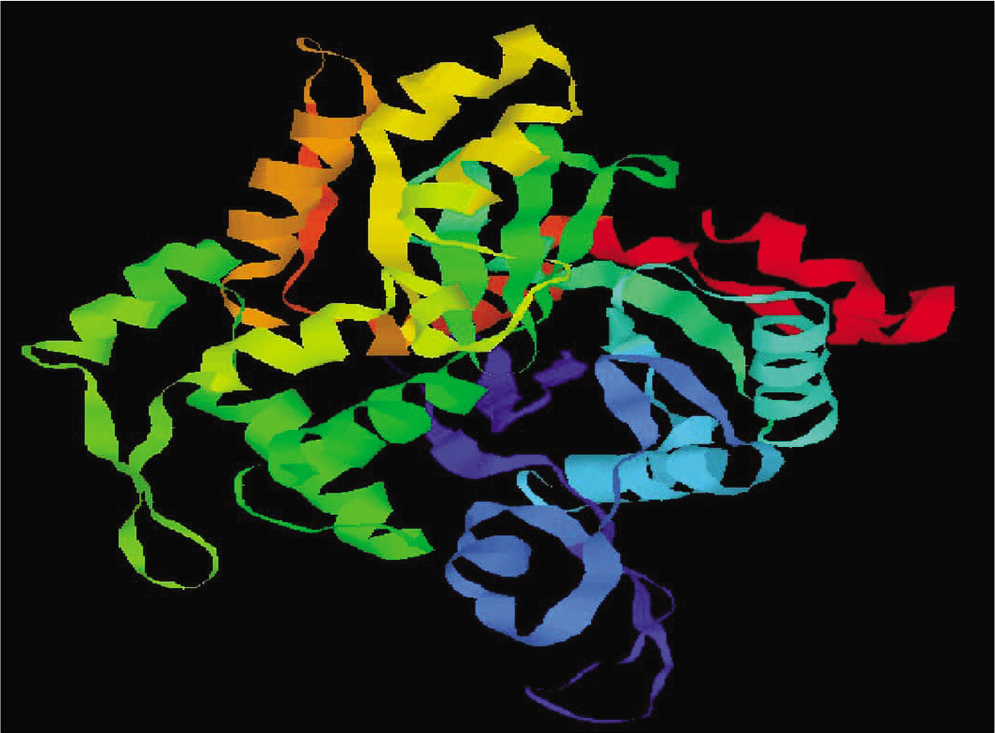
- Representation of modeled actin protein image of Leishmania donovani.
To check the resemblance between the secondary structure of model protein and template enzyme, we used the program called SOPMA and this program showed that the protein had 134 residues in a-helix (35.64%), 70 residues in extended strands (18.62%), 26 in b-turn (6.91%), and others (146, 38.83%) in random coil form. Hence, these results show that the secondary structure of both resemble to each other. The results after validation of refined model by Ramachandran plot analysis showed that model has 88.8% of residue in the most favored region and 10.2% in the additional allowed region [Figure 3]. By this, we conclude that no bad scores for main-chain or side-chain parameters. These quality check result says that selected structure of model has reliable conformation for protein docking.
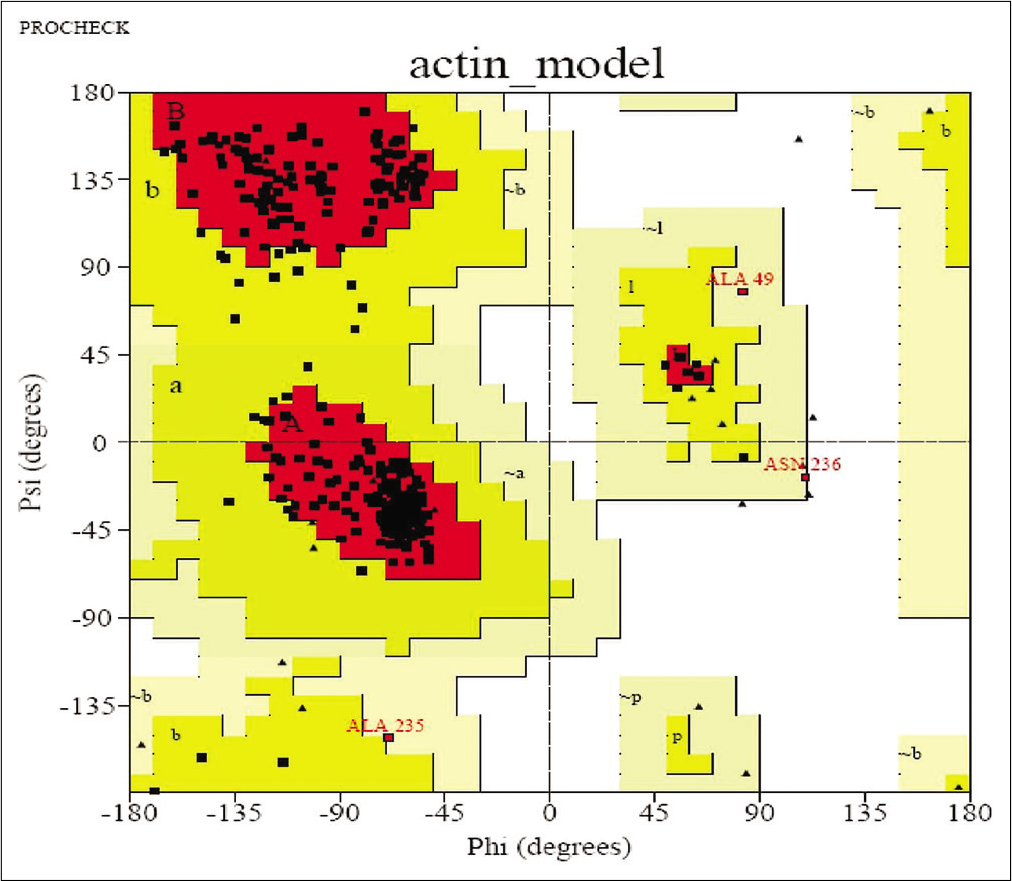
- Ramachandran Plot.
Docking studies
These studies were done for the predication of actin inhibitors. Docking between actin protein and different ligand including antileishmanial drugs and miscellaneous compounds was carried out in Ligandfit tool of discovery studio which provided the 11 different binding sites in actin model. These ligands were ranked on the basis of their dock score in discovery studio. All these compounds were then docked by GOLD and ranked on the basis of fitness score. Use of two docking methods helps in scoring of compounds based on the different scoring functions. It gives a result like consensus scoring. After comparing all the results from discovery studio and GOLD, the compound showing reasonable good score, i.e., high dock score by discovery studio and high fitness score by GOLD can be used as potential ligand.
These result from discovery studio shows that most compounds show good dock score. These compounds may be used as potential drugs. Almost ten compounds have a dock score above 70, as shown in [Table 1]. These results from Discovery studio also represented Graphically, as shown in [Figure 4]. Here hydrazide compounds, oxazole ring system containing compounds, serine protease inhibitor, and ATP analog showed the maximum Dock score. Now all these compounds were further used for docking with GOLD.
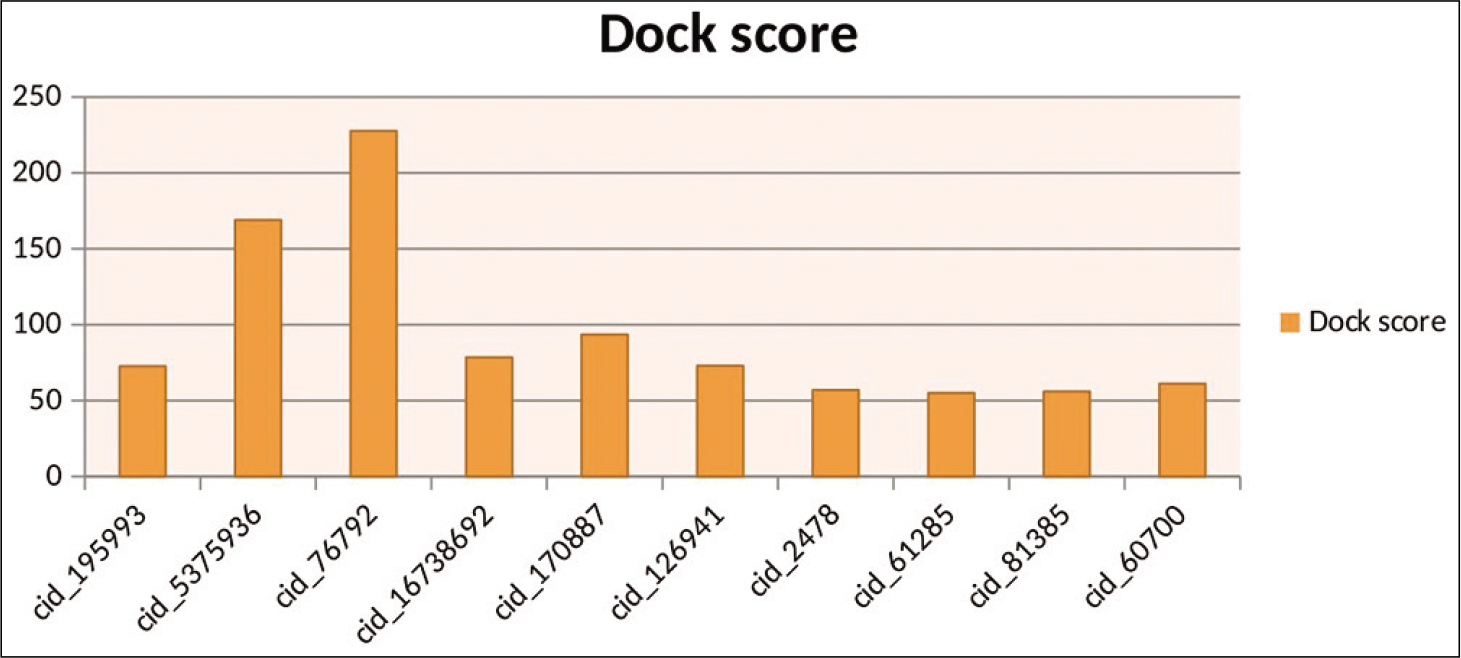
- Graphical representation of top ten results by discovery studio.
| S. No. | Compound id | Dock core | Amino acid involve in h bonding |
|---|---|---|---|
| 1. | cid_195993/Epdeatpasein | 72.63 | SER 15, GLY 16, LYS 19, ASP158, GLY159, VAL160, HIS162 |
| 2. | cid_5375936/1-(3,4-dimethyl-1,2-oxazol-5-yl)-2-[(E)-3-nitrosobut-2-en-2- yl] hydrazine | 168.948 | LYS 337, GLN138 ASP155, ARG336 |
| 3. | cid_76792/ 4-methoxybenzohydrazide | 227.65 | ASP155 |
| 4. | cid_16738692/Phosphatidylinositols | 78.529 | GLN60, LYS207, LYS214, LYS337 |
| 5. | cid_445420/latrunculin A | 41.982 | LYS207, TYR70, THR204 |
| 6. | cid_91557/methylene-ATP | 84.48 | SER15, GLY16, MET17, LYS19, ASP158, LYS207, LYS214, GLY159, HIS162, GLY303, GLU208 |
| 7. | cid_440317alpha-thioadenosine triphosphate | 86.32 | SER15, GLY14, MET17, LYS19, VAL160, HIS162, LYS214, GLY303 |
| 8. | cid_5957/adenosine triphosphate | 84.401 | SER15, LYS19, GLY16, ASP158, GLY159, LYS207, LYS214, LYS337 |
| 9. | cid_33113/gamma-imino-ATP | 81.03 | SER15, GLY303, MET17, LYS19, ASP158, LYS207, ARG211, LYS214 |
| 10. | cid_15993/de-oxy ATP | 76.09 | MET17, LYS19, ASP158, LYS207, ARG211, LYS214, GLY303 |
| 11. | cid_194049/ATP gamma benzylamide | 81.417 | SER15, LYS19, GLN60, ASP158, GLY303 |
Docking results of GOLD shows that compounds such as ATP analog, oxazole ring containing compounds, sulfonamide, and sulfonyl compounds and anticancer drugs (topotecan) have shown maximum fitness score, as shown in [Table 2]. The result obtained from ligand protein interaction by GOLD also represented Graphically, as shown in [Figure 5].
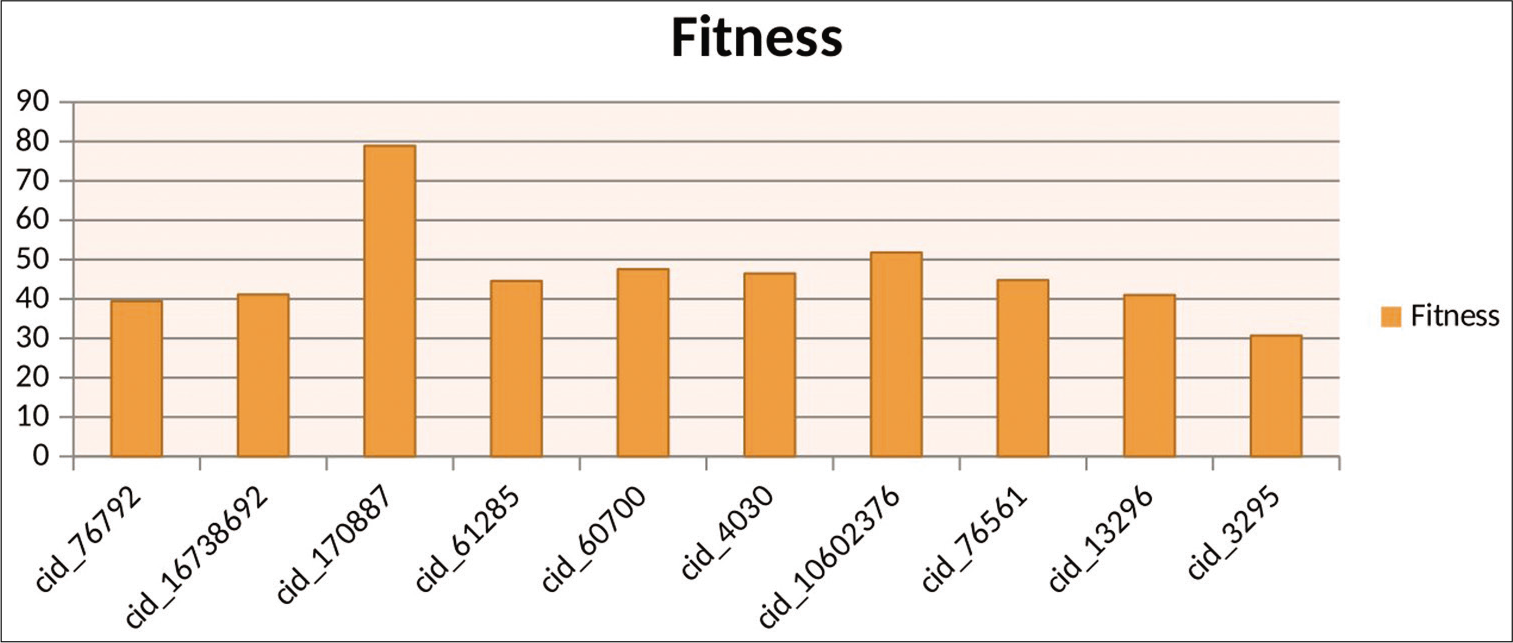
- Graphical representation of top ten results by GOLD.
| Compound | Fitness | S(hb_ex) | S(vdw_ex) | S(hb_in) | S(vdw_in) |
|---|---|---|---|---|---|
| cid_76792/4-methoxybenzohydrazide | 39.38 | 10.77 | 25.25 | 0.00 | ‒6.12 |
| cid_170887/aza-epsilon-ATP | 78.91 | 8.51 | 52.63 | 0.00 | ‒1.96 |
| cid_76561/benzylsulfonylbenzene | 44.79 | 0.00 | 34.96 | 0.00 | ‒3.27 |
| cid_27745/1-chloro-4-(2-chloroethylsulfonyl)benzene | 40.51 | 0.00 | 30.22 | 0.00 | ‒1.03 |
| cid_61285/N-Tosylbutylamine | 44.57 | 0.00 | 36.32 | 0.00 | ‒6.19 |
| cid_3295/ethoxzolamide | 30.72 | 0.00 | 38.28 | 0.00 | ‒4.05 |
| cid_4030/mebendazole | 46.46 | 0.00 | 37.79 | 0.00 | ‒5.50 |
| cid_60700/Topotecan | 47.55 | 0.00 | 40.91 | 0.00 | ‒8.69 |
| cid_10602376/2,4,5-tris(4-bromophenyl)-1,3-oxazole | 51.80 | 0.00 | 41.98 | 0.00 | ‒5.93 |
| cid_16738692/Phosphatidylinositols | 41.11 | 0.00 | 37.87 | 0.00 | ‒10.96 |
By comparing the result from both GOLD and discovery studio, we have observed that some compound such as hydrazide compound, oxazole ring containing compound, and anticancer drug such as topotecan and ATP analogue have shown good dock score by discovery studio but compounds such as sulfonyl compound, sulfonamide, oxazole ring containing compound, and ATP analogues and anticancer drug topotecan have shown good fitness score by GOLD. As oxazole ring containing compounds and topotecan and ATP analogs have shown good results from both GOLD and discovery studio, so we can use these compound as potential ligand.
DISCUSSION
Leishmania donovani actin is potential target for designing novel inhibitor for the treatment of visceral leishmaniasis. Keeping this in view, we have constructed a 3D model of the actin protein by Swiss model using the yeast actin-human gelsolin segment 1 complex (PDB ID: 1 yag) as a template. After loop refinement and side chain refinement, refined model structure was obtained. The refined model was then validated by Procheck and was found to be reliable. Then, the model was taken for docking studies.
Initially docking studies of L. donovani actin were done by Ligandfit tool of DISCOVERY STUDIO and the compounds with score more than 70 were then again docked by GOLD. These studies have shown that the compound such as oxazole ring containing compounds, Topotecan, and ATP analogs has greatest binding affinities to the active site of actin protein. The fitness score was compared with available antileishmanial drugs by GOLD and it was found that all the potent compounds have shown fitness score higher than available antileishmanial drugs.
CONCLUSION
This is the first report of ligand protein interaction for the treatment of leishmaniasis by acting on actin protein of L. donovani. However, inspite of existing drugs, new drugs for leishmania are still needed and for the designing of active inhibitors research gaps should be fulfilled.
Homology modeling of Actin protein of Leishmania give new light on the ligand binding features. The results favour the potential candidature of these compounds as lead molecule in designing future antileishmanial drugs research.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Functional aspects of the Leishmania donovani lipophosphoglycan during macrophage infection. Microbes Infect. 2002;4:975-81.
- [CrossRef] [Google Scholar]
- Leishmaniasis: Current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305-18.
- [CrossRef] [PubMed] [Google Scholar]
- A novel form of actin in Leishmania: Molecular characterisation, subcellular localisation and association with subpellicular microtubules. Mol Biochem Parasitol. 2004;134:105-14.
- [CrossRef] [PubMed] [Google Scholar]
- Actin molecular structure and function. Curr Opin Cell Biol. 1993;5:41-7.
- [CrossRef] [Google Scholar]
- Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433-73.
- [CrossRef] [PubMed] [Google Scholar]
- Searching for a function for nuclear actin. Trends Cell Biol. 2000;10:92-7.
- [CrossRef] [Google Scholar]
- Nuclear DNA helicase II/RNA helicase a binds to filamentous actin. J Biol Chem. 2002;277:843-53.
- [CrossRef] [PubMed] [Google Scholar]
- An actin-ribonucleoprotein interaction is involved in transcription by RNA polymerase II. PNAS. 2003;100:6475-80.
- [CrossRef] [PubMed] [Google Scholar]
- Functional cooperation between the microtubule and actin cytoskeletons. Curr Opin Cell Biol. 2000;12:63-71.
- [CrossRef] [Google Scholar]
- Conserved microtubule actin-interactions in cell movement and morphogenesis. Nat Cell Biol. 2003;5:599-609.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular links between endocytosis and actin-cytoskeleton. J Cell Biol. 2000;150:F111-6.
- [CrossRef] [PubMed] [Google Scholar]
- Pathways linking endocytosis and actin cytoskeleton in mammalian cells. Exp Cell Res. 2001;271:45-56.
- [CrossRef] [PubMed] [Google Scholar]
- An unconventional form of actin in protozoan hemoflagellate, Leishmania. J Biol Chem. 2008;283:22760-73.
- [CrossRef] [PubMed] [Google Scholar]
- Database resources of the national center for biotechnology information. Nucleic Acids Res. 2010;38:D5-16.
- [Google Scholar]
- Clustal W and clustal X Version 2.0. Bioinformatics. 2007;23:2947-8.
- [CrossRef] [PubMed] [Google Scholar]
- Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497-500.
- [CrossRef] [PubMed] [Google Scholar]
- Comparative Protein Structure Modeling with MODELLER Current Protocols in Bioinformatics. Hoboken: John Wiley & Sons, Inc.; 2006.
- [Google Scholar]
- SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11:681-4.
- [CrossRef] [PubMed] [Google Scholar]
- Modeling and molecular docking studies on Rnase Aspergillus niger and Leishmania donovani Actin: Antileishmanial activity. Am J Biochem Biotechnol. 2013;9:318-28.
- [CrossRef] [Google Scholar]
- In silico molecular modeling and docking studies on the leishmanial tryparedoxin peroxidase. Braz Arch Biol Technol. 2014;57:244-52.
- [CrossRef] [Google Scholar]
- Detailed comparison of the protein-ligand docking efficiencies of GOLD, a commercial package and ArgusLab, a licensable freeware. In Silico Biol. 2006;6:601-5.
- [Google Scholar]




